

文 | 財健道 嚴瑞
編輯 | 楊中旭
2021年12月24日,平安夜。信達生物(HK.01801)宣布國家藥品監督管理局(NMPA)正式受理信迪利單抗一新適應癥聯合用藥的上市申請。這大概是信達長達10天以來,第一次松了口氣。
此前,中國醫藥市場遭受重創,以致信達不得不發布公開聲明,以平息負面情緒——12月15日,英國《金融時報》稱美商務部將幾家中國生物技術公司列入實體名單,挫傷醫藥板塊股價;12月16日,FDA腫瘤學卓越中心(OCE)主任Richard Pazdur的幾句話,再次重創A/H股創新藥板塊。
中國創新藥國內“白菜價”、爭取“出海”盈利,是近階段最受推崇的發展邏輯。自1982年開放新藥快速通道后,美國FDA成為創新藥上市首要選項之一。不過,2014年以來,PD-1/PD-L1靶點的7種抗體,已經獲取超過85種腫瘤適應癥的審批,有評論稱:
“7種抗體的單抗/雙抗,與各種聯合治療方案、各類腫瘤適應癥之間不斷‘排列組合’,已經脫離了創新的初衷——也許之后,藥企自己的PD-1/PD-L1+xxx雙抗,就會先‘卷’起來。”
對此,相關專業人士評論:“排列組合的說法當然有些片面,但(藥企們)確實為了滿足各類替代終點而上市,投入了巨量的資源。”

從2016年開始,Pazdur就對企業對新適應癥“瓜分潮”表示不滿。作為FDA腫瘤學卓越中心主任、血液學和腫瘤學產品辦公室 (OHOP) 主任,直接負責領導開發、執行癌癥藥物及治療性生物制劑的綜合監管方法。
自2019年底,FDA開始出臺更明確的臨床試驗指南、重新審核以替代終點加速審批的腫瘤治療藥物。以“替代終點”上市,卻無法通過確證性審查的“空頭支票”被撤市——因為醫療領域承受不了一場泡沫化嚴重的“次貸危機”。
01 Pazdur一講話中國市場震三震
12月16日,FDA腫瘤學卓越中心(OCE)主任Richard Pazdur接受Prevision Policy采訪時的發言,給創新藥的傷口“撒了把鹽”:

“新藥獲批僅靠一個國家的臨床數據,比如中國,顯然是有問題的——這和美國在臨床試驗中努力增加患者多樣性的原則背道而馳。”
Pazdur不僅是FDA創新藥審批制度的“一把手”,還與中國創新藥市場關系匪淺。
2018年,Pazdur曾“敦促”中國將合格的低價PD-1/PD-L1抑制劑引入美國市場。那年4月訪問上海浦東張江高科技園區時,他在采訪中表示:只要質量好,FDA一定會接受僅依靠中國產生臨床數據產生的申請;雖然不會以價格作為標準,但歡迎價格低廉的產品。
在美國癌癥研究協會(American Association for Cancer Research)一次名為“東方與西方相遇:中國制藥探索西方市場”的會議上,Pazdur在問答環節講話時也表示:“中國公司赴美競爭PD-1價格,可能對所有人都是件好事——目前為止,我們沒有看到西方主要制藥公司在價格上做出改變”。
“我可以看到一個非常簡單的開發戰略,”他說,“例如在肺癌方面,你可以簡單地做一些大型制藥公司已做過的研究。已經知道效應值(effect size),甚至都不必做非劣效研究就能上手,統計計劃寫起來也容易——你甚至都不需要成為一名統計學家。”
他還認為,中國對FDA已批準藥物的模仿,也將順利獲批:“很明顯,它們會產生非常相似的結果,所以我們在批準這類藥物方面,幾乎沒有什么可說的。”
此前這些表述,與其接受Prevision采訪的發言,似乎背道而馳。近日報道中Pazdur的觀點有三:
1.不能只做單一國家單一人種臨床;
2.“me-too”靶點泛濫,應開展國際化管理;
3.PD-1/PD-L1同質化競爭嚴重(針對國際頭部創新藥企)。
前兩點,無疑會引發中國創新藥市場巨震。因為中國腫瘤免疫治療基本維持在“me-too”水平,且多項臨床試驗都依靠中國患者數據進行驗證。此前,百濟神州(688235)澤布替尼成功出海、近日CDE宣布擬將其納入突破性療法,無疑又助長了這一邏輯。
作為主要依靠中國臨床數據進行申請的種子選手之一,信達的信迪利單抗首當其沖。在隨后發布的會議紀要中,信達詳細解釋了亞組患者入組率問題:盡管中國患者的入組比例在四種藥物中最高,但仍在臨床指南合理范圍內,與FDA要求臨床試驗多中心的規則不沖突;同時,晚期患者的入組率在四種藥物中排名第二高,也更符合真實世界情況。
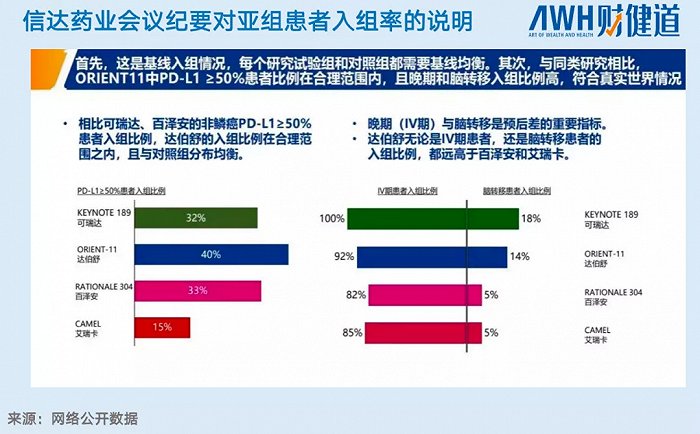
不過,針對這一藥物是否稱得上“the best”的爭論沒有停止。有醫學相關人士表示:信迪利沒有在各替代終點風險指標上超過默沙東K藥(Keynote189)。
因為普遍缺乏頭對頭試驗,所以腫瘤藥物臨床試驗,多對患者入組后總生存期(OS)、腫瘤得到控制的無進展生存期(PFS)等數據建立分析模型,來預測治療方案的有效性。
模型所得出的風險比(HR)數據越小,代表患者從中獲益可能性就越大。比如,PFS試驗HR=0.76,就代表患者入組無進展期死亡率下降24%。普遍來說,患者PD-L1表達越高,免疫治療效果就越好。在臨床試驗中,低表達率比如PD-L1<1%的患者的臨床HR數據,便可以作為“the best”的證據之一。

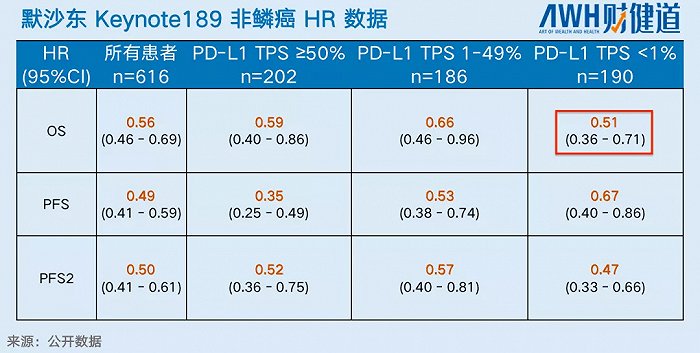
會議紀要中,信達生物使用的K藥非鱗癌PD-L1<1%PFS HR數據為0.68,信迪利(即達伯舒)的這項數據為0.598,顯示信迪利獲益率更佳。

接著,在OS HR數據方面,信迪利單抗似乎也排名第一,為0.60,好于默沙東K藥的0.608。然而,美國臨床腫瘤學會(ASCO)年會2020年5月公布的K藥數據中,其OS HR為0.51,遠超信達生物會議紀要中取用的數據。僅就這一數據,信迪利就很難論證所謂的“best”。
中國創新藥出海邏輯,于是陷入一場爭論不休的“羅生門”。
02 FDA只針對中國藥企嗎?
當然不是針對中國。FDA針對的是任何鉆臨床試驗替代終點的空子、“以次充好”以謀取利益的企業。若“以次充好”的泡沫泛濫,醫療領域絕難承受一場“次貸危機”式的災難。
而藥企們之所以敢于自稱“me-better”或“the best”,和FDA審批制度有關。上世紀80年代開始,FDA開始使用替代終點和加速審批,來幫助腫瘤治療藥物上市,打開了各種風險分析路徑的可能性。
盡管普遍來說,PD-L1表達率越高的患者,使用免疫治療的有效性應當越大。但免疫起效機制并不僅由一項表達率標準就能確定,表達率90%但免疫治療無效的案例、表達率1%但單藥治療療效就不錯的案例都不少。據業內相關人士介紹,某種程度上,PD-L1表達率并完全是臨床判斷的科學標準,還是藥企借以上市所使用的預測模型維度之一。
替代終點不可能真的替代臨床終點,就好像當年的信貸危機,泡沫永遠不能代替真正的收益。比如說,腫瘤縮小當然可以作為替代終點,支持藥品上市,但它是否能提高病人的總生存率,仍需確證性實驗來驗證。好比狼人殺里有“金水”“銀水”之別,何況“金水”也并非永遠正確。
因此,作為以“可能性”上市的藥物,只要達到一定水平,在沒有真實數據和確證性試驗,各藥總能找出一款分析模型來自證有效。
既然效果不確定,那為什么還要支持藥物加速審批上市呢?因為腫瘤治療藥物研發太難了。若以臨床試驗終點的“金標準”、即患者的總生存率為唯一標準,那么,腫瘤治療藥物通過審批就會極為困難。
上世紀末FDA就認識到,在嚴重或罕見疾病方面,藥少、試驗少、審批少很可能造成惡性循環。因此,讓患者盡可能有藥可用,成為創新藥使用替代終點、加速審批上市的起點。
加速和替代,配合腫瘤治療藥物的巨大市場,必然帶來藥物扎堆現象。于是,Pazdur自2016年也就是第一款PD-1/PD-L1藥物上市兩年開始,就開始對以美國為首的腫瘤創新藥企表達不滿。
那時Pazdur所謂PD-1/PD-L1同質化競爭,直指頭部藥企比如默沙東、羅氏、百時美施貴寶等,在單一靶點免疫治療耗費了太多資源。2019年以前,他不僅沒有針對中國藥企,反倒在“敦促”中國藥企仿制創新藥來平衡創新藥市場價格。
然而,Pazdur理想中的低廉價格、創新導向并未實現。中國創新藥企不僅沒有降價,還都指望著藥物能“出海”撈回國內醫保談判里被打下去的利潤。
2021年12月15日,也即接受Prevision采訪前一天,他和同事Julia Beaver在《新英格蘭醫學》雜志上發表《檢查點抑制劑開發的狂野西部》一文,繼續表達了這種失望情緒。

“過去7年中,FDA已經批準7種針對PD-1/PD-L1通路的抗體,及這類藥物超過85種的適應癥。”[1]文章開篇劍指的7年前,即是2014年默沙東K藥、百時美施貴寶O藥獲批的那個時間點。
截至2021年8月,據相關媒體統計。出去這已經獲批的適應癥,在ClinicalTrials.gov官網搜索“PD-1”,仍有2903項開展中的相關臨床試驗,以及1343項仍在招募中的臨床試驗。
幾千個研究中,新適應癥或突破性療法很少,大部分都是7種已有抗體成分藥物的聯合治療方案,可以理解為已有抗體藥物與不同適應癥的“排列組合”。
Pazdur認為,這大大降低了癌癥免疫治療研究投入的邊際效應。一來,沒有研究來對比不同療法的可及性與經濟性;二來,盡管這種藥物的設計、治療方案確定已然“流水化”,但新“排列組合”方案的研發、審批支出仍居高不下。相當于做著“仿制藥”的工作,卻投入“原研藥”所需的資源、掙著“原研藥”的錢。
在2016年第52屆美國臨床腫瘤學會(ASCO)年會上,Pazdur就表示,“人們應該問一下他們自己……我們是不是應該更好地使用那些資源,用來開發更多創新藥物?”。
基于此,才有“東方與西方相遇”大會上,Pazdur的發言。他直接表示,非常歡迎中國公司“仿制”PD-1/PD-L1打破美國“狂野西部”同質化競爭、不愿降價的僵局。
然而,同質化競爭并非某一個國家的特殊缺陷。“狂野西部”文章中所抨擊的現象——比如贊助商一擁而上,基于非隨機、單臂試驗制定批準策略、招募醫療需求未得到滿足的患者以獲得加速批準等等——在世界范圍內(當然包括中國),都已經并也許仍將頻繁出現。
至此,就算完全拋開對藥物研究的數據分析不談,我們也能明白Pazdur幾句反同質化、注重臨床數據有效性的話,為何能動搖市場對中國創新藥企的信心:
對于中國創新藥市場來說,“me-too”仍是大多數企業的天花板,大多數競爭者都是已知“內卷”進入白熱化階段、卻還不撤退的人。他們明白,想在“次貸危機”中牟利,當然要迎接一場豪賭。
03 FDA審批收緊確證性審查“秋后算賬”
也許,前幾年確實是PD-1/PD-L1的紅利期,但快速審批機制從來不是為投機者準備的捷徑。
2012年2月,當時的FDA局長Echenbach在《華爾街日報》刊登《FDA的藥品審批應基于安全性,有效性應留給上市后研究》一文,主張改革藥品監管,從而達到鼓勵創新、降低藥價、造福患者的目的。這意味著,當時的FDA對腫瘤治療藥物市場,持鼓勵、觀望態度。
畢竟,想要創新,就要一定程度上允許“泡沫”的存在,好比鄧小平“開窗通風、蒼蠅也會進來”的比喻。所以,Pazdur2016年開始對創新藥的批評,完全不被藥企高管們認同。
浸淫創新研發工作動輒幾十年的藥企們,深知創新之難,它不僅需要資源的堆砌,更需要機運的青睞。他們中的大多數人認為,已有靶點下不同抗體藥物之間的聯合治療、即“排列組合”,才是真實世界的癌癥免疫治療趨勢。從2014年至今,現實映證了他們的說法。
前文中針對PD-1/PD-L1的7種抗體藥物,能夠取得超過85項適應癥審批,依靠的是替代終點和加速審批。這是所謂紅利期的來源——自1982年以來,快速審批通道大多為腫瘤治療藥物“占領”;反之,腫瘤治療藥物也鮮少不在快速審批行列內。
但是,FDA 在2018年《美國醫學會雜志》腫瘤學分冊發表的一項研究中公布,對93 個獲得加速批準的藥物,僅有55%上市后證實了其獲益;獲得加速批準的適應癥,僅有40%完成了確證性臨床試驗。
2019 年發表在《美國醫學會雜志·內科學》的一項研究[3]發現,FDA 通過使用替代終點加速審批的癌癥藥中,只有20%在確證性試驗中提高了總生存率。該研究的作者在2017年的一項研究中還說明道[4],通過快速審評批準的藥物,使藥品標簽上的安全性警示變更率增加38%。
一段時間的試驗后,FDA開始以監管的“有形之手”,去修整腫瘤免疫藥物市場的“無形之手”。2019年12月開始,FDA開始出臺針對性指南方案,開展對替代終點上市的腫瘤治療藥進行數據復核,以“慢撒氣”的方式,緩慢展開“秋后算賬”攻勢。而宏觀也好,產業也罷,慢撒氣,不急轉彎,是監管水準高低的標志。
2019年12月20日,FDA發布名為《證明人用藥物和生物制品具有有效性的實質證據》(DemonstratingSubstantial Evidence of Effectiveness for Human Drug and Biological Products)的行業指南。
2020年11月,FDA高級官員在預防政策/癌癥研究之友生物制藥大會上證實,將對加速審批藥物開展審查。
2021年1月25日, FDA 藥品審評與研究中心(CDER)發布藥品方面的指南制定計劃,其中《滿足基于一個充分且良好對照的臨床研究以及確證性證據的實質性證據標準》指南,脫胎于2019年12月指南草案 IV.B 小節的標題。
3個月后,FDA正式宣布,在4月27日至29日舉行腫瘤藥專家咨詢委員會(ODAC)會議,討論三個癌癥免疫療法PD-1/PD-L1藥物獲得加速批準的 6個未確證臨床獲益的適應癥的未來。《聯邦公報》通告表示,此次會議是由腫瘤卓越中心(OCE)要求召開的,是其“對整個行業范圍腫瘤產品方面確證性試驗未證實臨床獲益的加速審批的評估”的一部分。
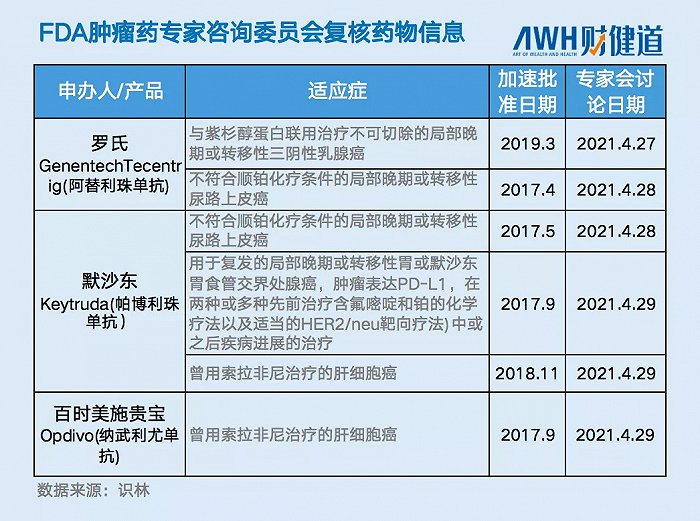
面臨審查和撤銷風險的藥品包括Genentech阿替利珠單抗的兩個適應癥,默沙東的帕博利珠單抗的三個適應癥,以及百時美施貴寶的納武利尤單抗的一個適應癥。
此前,3月早些時候,百時美施貴寶、阿斯利康、默沙東和羅氏四家公司其實已經與FDA達成協議、宣布撤銷了另外四個新適應癥的加速審批,撤銷原因是確證性試驗不符合上市后要求(PMR)。
6月18日,FDA 發布首份患者體驗數據在監管決策中使用的評價報告。作為 2016 年《21 世紀醫藥法案》的一部分,第 3004 節要求FDA 需要在 2021 年、2026 年和 2031 年分別發布患者數據評價報告。
報告指出,患者體驗數據應該成為患者報告結局(PRO)和臨床結局評價(COA)的核心作用。這代表著,根據替代終點加速審批上市的腫瘤治療藥物們,將被要求進行上市后的確證性實驗,并隨時面臨撤市風險。
2021年12月8日,FDA發布了《使用真實世界數據和真實世界證據支持藥品和生物制品監管決策的考量因素》指南草案。當然,都是為了在藥品上市后能繼續觀察、研究,且最重要也最關鍵地——進行監管。
04“秋后算賬”,但有分寸
中國藥企能否真的如Pazdur所說,以大量低價打入美國市場?只要免疫治療還處在技術前沿,就很難。
FDA“有形的手”再收緊,也不能干預市場價格,尤其是對創新藥市場。從K藥、O藥上市至今,腫瘤免疫治療藥物價格之昂貴,未有變動。
FDA心臟病、血液、內分泌和腎臟病學辦公室主任Ellis Unger認為,假設一個藥具有臨床獲益的可能性為 70%,那么該路徑中獲批的藥物中有30% 就應該退出市場。但想要求藥物撤市,實際上很難,因為FDA無法主導市場決策。
某種程度上,確證性審查大會、撤市和新指南的出臺,都是FDA應對市場“內卷”問題作出的專業性努力。圍繞專業性做文章,基本是其能做出的最大努力。
對FDA來說,只有在保證專業性的情況下,新藥才可申請加速批準。從1982年開始設立新藥加速審批途徑至今,FDA藥物審評時間從20世紀80年代審評時間接近3年,下降到現在的1年左右,效果顯著。不過,創新藥研發從臨床試驗到正式批準上市的總平均時間,并無顯著變化,也就是說開發難度不降反增。[5]
越快速的審批通道,對藥物臨床數據的要求也越高。比如突破性療法認證(即BTD),要求藥物在一開始臨床試驗階段就證明,藥物能在一個或多個有臨床意義的指標上,較現有療法有顯著改善。如果一個藥物申請突破性藥物失敗,FDA不會將取消其快速通道認定程序,研發單位需要重新申請。
迄今為止,百時美施貴寶的Opdivo獲得10項BTD認證,默沙東的Keytruda則獲得9項BTD認證,領跑群雄。但FDA對上述藥物的確證性審查并未“仁慈”。上文審查大會中審核的6個適應癥中,有2個已經被撤銷。
所以,盡管工商業界對FDA的監管有不同看法,但對其專業能力卻一致認同。許多FDA審查員是碩士或博士學位,擁有“醫生、律師、分析師、科學家”等多重身份,或在大學或科研機構兼任教授。
默克公司的一位負責人曾表示:“我們雖然不一定認同FDA的決定,但我們一定尊重FDA的決定。”
當然,就算沒有確證性審查趨嚴,藥企名正言順的“排列組合”戲法,也已經要玩到極限了。
以信達生物此次頗受爭議的信迪利單抗為例,其申請的是信迪利單抗聯合培美曲塞和鉑類用于非鱗狀非小細胞肺癌(NSCLC)的一線治療。
這個適應癥簡直是大中之大——首先,肺癌是全球死亡率最高的惡性腫瘤,發病率也是世界第二;其次,非小細胞肺癌患者大約占全部肺癌患者的80%至85%;最后,美國2021年全年超250億美元的PD-1銷售額中,約有60%來自非小細胞肺癌適應癥,且主要是在一線治療。
以再生元為例。其Libtayo同樣申請了在非小細胞癌癥一線治療為例,它們需要對抗的是默沙東、百時美施貴寶和羅氏。據業內人士轉載,華爾街著名分析機構SVB Leerink分析師預測,Libtayo即使獲批非小細胞肺癌一線,其在所有PD-1/PD-L1市場中的份額永遠不會超過5%。
飽和狀態下,市場自有其生態模式。越廣譜、越早上市的藥品(如默沙東K藥)占商業保險支付比例越大,與保險支付方的關系也更緊密。廣譜免疫治療藥物往往能提供更高比例的回扣,形成“回扣墻”,在產業鏈中下游均形成某種程度上的壟斷。
盡管包括恒瑞在內一眾中國創新藥企都選擇從大適應癥入手,但這也許并非好事。想要與占盡先機的廣譜免疫治療藥物競爭,除開價格壁壘不談,在臨床頭對頭試驗中證明“me-better”甚至“the best”其實相當困難。
而冷門適應癥沒有成熟案例可復制,做起來可能更難。這種情況下,除了追求“the best”,“me-too”甚至“me-worse”看不到未來,所謂的價格戰便無從談起。
“中國目前大多處于中度創新的創新藥企,還是要擁抱國內市場。”一位醫藥板塊分析師表示,盲目“出海”的下場只能是“賠本賺吆喝”。可中國的藥物評審和審批制度也趨于嚴格。
2021年7月2日,國家藥監局藥品審評審批中心(CDE)下發《化學藥品創新藥上市申請前會議藥學共性問題相關技術要求(征求意見稿)》和《以臨床價值為導向的抗腫瘤藥物臨床研發指導原則(征求意見稿)》,特別針對腫瘤藥物提出與最新治療方案的頭對頭臨床試驗,并闡明產品商業化差異性。
如此看來,中國創新藥企好像進退兩難起來。但這個“難”,本就是創新的奧義。
中國創新藥“me-too”困境,是冰凍三尺、非一日之寒——企業不改把創新扭曲成看似容易的仿制熱,因為“次貸危機”的泡沫終會破滅。原國家食品藥品監督管理總局局長畢井泉告訴《財健道》:“需要把評審中心(CDE)做大做強,鼓勵真正的創新。”
這需要長期耐心的大量投入,其中包含制度、資金和社會理念等多方面涵養。持續多年,才有可能找到一棵真正創新的萌芽。
(作者系《財經》研究員)
-END -
參考資料【1】Beaver JA, Howie LJ, Pelosof L, et al. A 25-Year Experience of US Food and Drug Administration Accelerated Approval of Malignant Hematology and Oncology Drugs and Biologics: A Review. JAMA Oncol. 2018;4(6):849–856. doi:10.1001/jamaoncol.2017.5618【2】Julia A. Beaver,M.D.,and Rechard Pazdur,M.D.:The Wild West of Checkpoint Inhibitor Development.(https://www.nejm.org/doi/full/10.1056/NEJMp2116863)【3】Gyawali B, Hey SP, Kesselheim AS. Assessment of the Clinical Benefit of Cancer Drugs Receiving Accelerated Approval. JAMA Intern Med. 2019;179(7):906–913. doi:10.1001/jamainternmed.2019.0462【4】Mostaghim S R, Gagne J J, Kesselheim A S. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study BMJ 2017; 358 :j3837 doi:10.1136/bmj.j3837
【5】柏林,范平安,史錄文,陳敬.從美國1985—2019年新藥批準情況看新藥研發和審批趨勢[J].中國新藥雜志,2021,30(20):1830-1835.